What Are the Different Classes of Medical Devices?
Medical devices play a critical role in modern healthcare, ranging from simple disposable products to complex implantable technologies. To ensure patient safety and appropriate regulatory control, medical devices are classified into different risk-based categories. Understanding medical device classes is essential for manufacturers, suppliers, and healthcare professionals, as classification determines regulatory requirements, conformity assessment procedures, and market access.
Why Are Medical Devices Classified?
Medical device classification is based on the level of risk associated with the device’s intended use. The higher the potential risk to the patient or user, the stricter the regulatory controls. Classification helps regulatory authorities ensure that medical devices placed on the market meet safety, performance, and quality standards. While classification systems may vary slightly between regions (such as the European Union, the United States, or other international markets), they generally follow a similar risk-based logic.
Medical Device Classes in the European Union
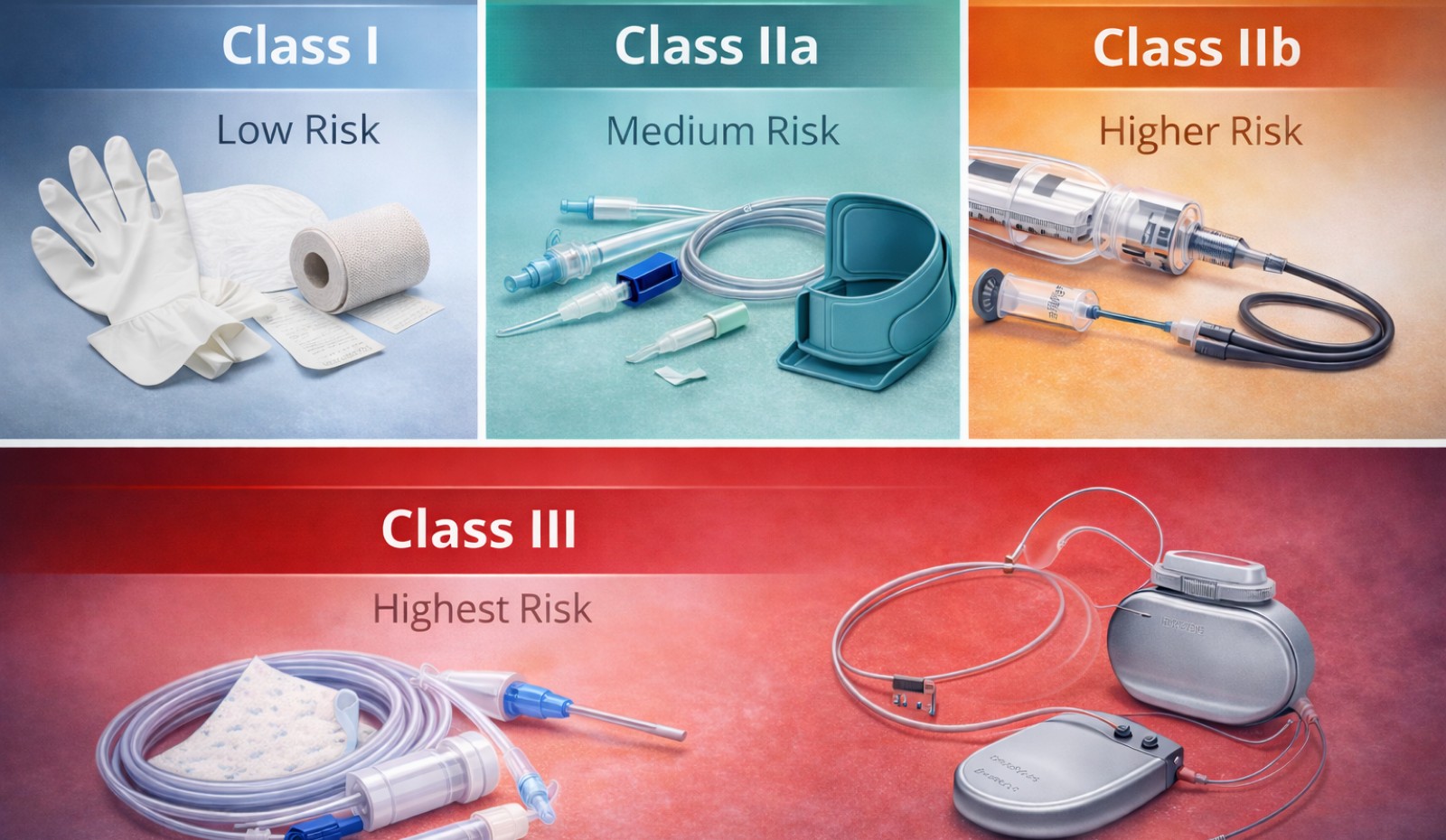
Under the EU Medical Device Regulation (MDR 2017/745), medical devices are divided into four main classes: Class I, Class IIa, Class IIb, and Class III.
Class I – Low Risk Medical Devices
Class I devices are considered low risk and typically non-invasive. Examples include bandages, surgical gloves, non-sterile tubing, and simple medical accessories. Many single-use plastic medical devices fall into this category when they do not come into contact with critical organs or the circulatory system. Manufacturers of Class I devices often self-certify compliance, although additional requirements apply for sterile or measuring functions.
Class IIa – Medium Risk Medical Devices
Class IIa devices present a moderate level of risk and are often invasive for short-term use. Examples include catheters, suction devices, infusion sets, and some diagnostic equipment. For Class IIa devices, manufacturers must involve a notified body to assess conformity. This class includes many disposable medical devices used in routine clinical procedures.
Class IIb – Higher Risk Medical Devices
Class IIb devices are associated with a higher risk, often due to long-term use or interaction with vital bodily functions. Examples include long-term invasive catheters, infusion pumps, and certain surgical instruments. Stricter clinical evaluation and regulatory oversight are required for Class IIb devices, particularly for single-use products designed for prolonged contact with the body.
Class III – Highest Risk Medical Devices
Class III devices pose the highest risk and typically include implantable or life-sustaining devices such as heart valves, implantable pacemakers, and neurostimulators. These devices require extensive clinical evidence, rigorous testing, and full involvement of notified bodies before they can be placed on the market.
Medical Device Classes in the United States
In the United States, the FDA uses a three-class system: Class I (low risk), Class II (moderate risk), and Class III (high risk). While the structure differs slightly from the EU system, the underlying principle remains the same: higher risk means stricter regulatory control.
Why Classification Matters for Medical Device Manufacturers
Correct medical device classification is crucial for regulatory compliance, time-to-market, and cost control. For manufacturers of single-use plastic medical devices—such as connectors, tubing, adapters, or stopcocks—classification directly impacts material selection, sterilisation methods, documentation, and quality management systems. At Promepla, understanding regulatory requirements and device classification is an integral part of supporting customers throughout the development and industrialisation of medical devices.
Latest news

November 6, 2025
Customers IFU

Our websites
